When you pick up a generic pill at the pharmacy, you expect it to work just like the brand-name version. But how do regulators know it’s truly the same? The answer lies in pharmacokinetic studies-the most widely used method to prove that a generic drug behaves the same way in the body as its branded counterpart. Yet calling them the "gold standard" is misleading. They’re not perfect. They’re not always enough. And for some drugs, they’re downright inadequate.
What Pharmacokinetic Studies Actually Measure
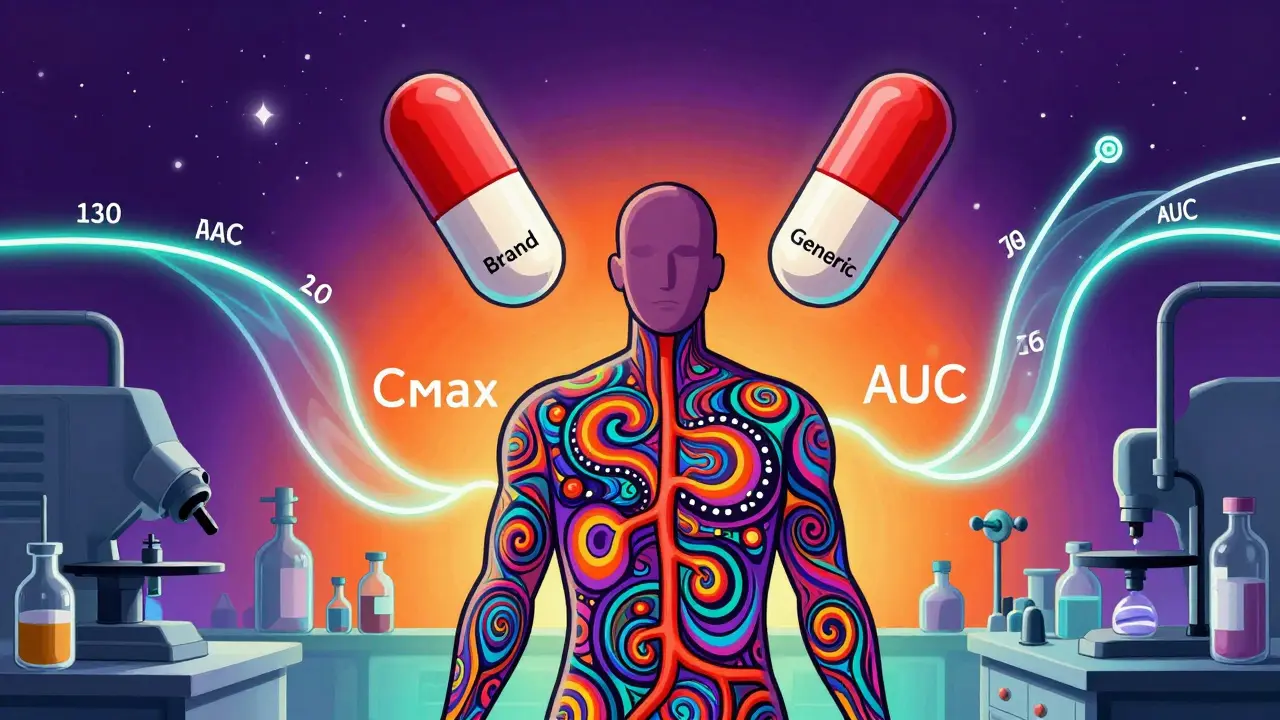
Pharmacokinetic studies track how a drug moves through the body: how fast it’s absorbed, how high it goes in the blood, and how long it stays there. The two key numbers they measure are Cmax (the highest concentration reached in the bloodstream) and AUC (the total exposure over time). These aren’t just lab curiosities-they’re direct indicators of whether the drug will have the same effect.
The FDA requires that the 90% confidence interval for the ratio of these values between the generic and brand-name drug falls between 80% and 125%. That means if the brand drug hits a Cmax of 100 ng/mL, the generic can range from 80 to 125 ng/mL and still be considered equivalent. For most drugs, that’s wide enough to account for normal human variation without risking safety or effectiveness.
These studies are done in healthy volunteers-usually 24 to 36 people-in a crossover design. Each person takes both the generic and the brand-name version at different times, with a washout period in between. This eliminates individual differences in metabolism from skewing the results. The tests are run under both fasting and fed conditions because food can dramatically change how a drug is absorbed.
Why It’s Not Really a "Gold Standard"
The FDA doesn’t call pharmacokinetic studies a "gold standard." They call them a "fundamental principle." That’s not just semantics. It’s a warning.
Therapeutic equivalence means the drug works the same in the body-not just that it reaches the same blood levels. For most drugs, blood levels are a reliable proxy. But not for all. Take narrow therapeutic index (NTI) drugs like warfarin, phenytoin, or digoxin. These drugs have a tiny margin between a helpful dose and a dangerous one. For them, the 80-125% window is too loose. The FDA now requires tighter limits: 90-111% for Cmax and AUC. Even then, there are cases where two generics with identical pharmacokinetic profiles still cause different clinical outcomes.
One 2010 study in PLOS ONE found that two generic versions of gentamicin, both meeting all regulatory requirements and showing identical in vitro dissolution profiles, produced significantly different effects in patients. The active ingredient was the same. The excipients were within tolerance. The blood levels matched. But the clinical response didn’t. That’s not a failure of the study-it’s a failure of assuming blood levels tell the whole story.
Where Pharmacokinetic Studies Fall Short
For oral tablets and capsules, pharmacokinetic studies work well. But for complex formulations? Not so much.
Take topical creams, ointments, or inhalers. You can’t measure drug levels in the blood the same way you can with a pill. A steroid cream might work locally on the skin, but barely enter the bloodstream at all. Measuring plasma concentration tells you nothing about whether it’s actually working where it’s supposed to.
That’s why regulators are turning to alternatives. Dermatopharmacokinetic methods (DMPK) use tape-stripping to measure drug concentration in skin layers. In vitro permeation testing (IVPT) uses human skin samples to see how the drug moves through layers. One study showed IVPT was more accurate and less variable than clinical endpoint trials for semisolid drugs. And it’s cheaper, faster, and doesn’t require human subjects.
Same goes for modified-release tablets. A tiny change in the polymer coating can delay release by 30 minutes, which might not show up in a standard AUC measurement but could mean the drug doesn’t last as long during the night. These subtle differences are invisible to traditional pharmacokinetic studies.

The Cost and Complexity Behind the Scenes
Running a single bioequivalence study isn’t cheap. It costs between $300,000 and $1 million-and takes 12 to 18 months from formulation to approval. That’s why generic manufacturers often delay development until the patent expires. It’s also why so many companies get stuck on Pitfall #2: proving bioequivalence for complex formulations.
Even when the active ingredient is identical, excipients (fillers, binders, coatings) can change how the drug dissolves. A change in particle size, a different grade of lactose, or a slightly altered coating can alter absorption. The FDA has issued 1,857 product-specific guidances as of 2023, each tailored to a particular drug’s unique behavior. There’s no one-size-fits-all rule.
Some manufacturers use the Biopharmaceutics Classification System (BCS) to avoid human studies entirely. If a drug is highly soluble and highly permeable (BCS Class I), and the formulation is similar, the FDA may waive the bioequivalence study. But that only applies to about 15% of drugs. For the rest, it’s back to the lab-and the volunteers.
Global Differences and Regulatory Gaps
The FDA and the European Medicines Agency (EMA) don’t always agree. The EMA tends to use a more rigid, one-size-fits-all approach. The FDA allows flexibility based on the drug’s properties. This creates headaches for global manufacturers trying to get approval in multiple countries.
And then there’s the rest of the world. Around 50 national regulators follow WHO guidelines, but implementation varies wildly. In some countries, studies are done with small sample sizes, poor controls, or outdated methods. That’s why you’ll sometimes see generic drugs with identical names but wildly different performance across borders.
Efforts like ICH M13A aim to harmonize standards, and 35 countries have adopted it. But harmonization doesn’t mean uniformity. It just means everyone’s using the same rulebook-sometimes reading different chapters.
The Future: Beyond Blood Levels
The field is changing. Physiologically-based pharmacokinetic (PBPK) modeling is now accepted by the FDA to support bioequivalence waivers for certain BCS Class I drugs. Instead of testing in people, scientists simulate how the drug behaves in a virtual human body using data on anatomy, enzyme activity, and blood flow. It’s not science fiction-it’s already being used.
For topical drugs, dermatopharmacokinetic methods are proving powerful. One 2019 study showed they could detect differences in bioavailability with over 90% accuracy-far better than clinical trials that require hundreds of patients.
Even in vitro tests are getting smarter. For some immediate-release drugs, dissolution testing with advanced models can predict in vivo performance better than human studies. PLOS ONE published research in 2010 showing that for certain formulations, lab tests were more reliable than human pharmacokinetic trials.
The message isn’t that pharmacokinetic studies are obsolete. They’re not. They’re still the backbone of generic approval. But they’re no longer the whole story.
What This Means for Patients
For most people, generic drugs are safe, effective, and life-changing. They save billions of dollars every year. But if you’re on a narrow therapeutic index drug, or you’ve noticed a change in how a generic works for you, it’s not just in your head.
Regulators are aware. That’s why they’re investing in better tools. But until those tools become standard, patients should report any unexpected side effects or loss of effectiveness after switching generics. That data feeds back into the system and helps improve standards.
The truth is simple: pharmacokinetic studies are the best tool we have-for now. But they’re not perfect. And the goal isn’t just to prove equivalence on paper. It’s to make sure every pill, no matter who made it, works the same way in your body.
Are pharmacokinetic studies required for all generic drugs?
No. For some drugs, especially those classified as BCS Class I (highly soluble and highly permeable), regulators like the FDA may waive human pharmacokinetic studies if the formulation is similar to the brand and dissolution profiles match. In vitro testing and modeling can sometimes replace human trials. But for most drugs-especially complex or narrow therapeutic index ones-pharmacokinetic studies are still mandatory.
Why do some people say generic drugs don’t work as well?
In rare cases, patients report differences after switching to a generic, especially with drugs like warfarin, thyroid hormone, or seizure medications. This isn’t always due to poor quality-it can be because of small differences in how the drug is released or absorbed. Pharmacokinetic studies may miss these subtleties, especially if the drug acts locally (like inhalers or creams) or if the patient’s metabolism is unusually sensitive. Regulatory agencies are improving methods to catch these cases, but patient feedback remains critical.
How long do pharmacokinetic studies take to complete?
A typical study takes 12 to 18 months from formulation development to final report. The actual clinical phase-where volunteers take the drugs-lasts about 4 to 8 weeks. But preparation, regulatory submissions, data analysis, and reporting add months. For complex drugs, the timeline can stretch longer due to multiple test conditions (fasting, fed, multiple doses).
Can a generic drug be approved without human testing?
Yes, under specific conditions. If a drug is BCS Class I, has the same formulation as the reference product, and passes rigorous in vitro dissolution testing, regulators may approve it without human pharmacokinetic studies. The FDA has approved dozens of generics this way. But this only works for simple, well-understood drugs-not for modified-release, topical, or injectable products.
What’s the difference between pharmaceutical and therapeutic equivalence?
Pharmaceutical equivalence means two drugs have the same active ingredient, strength, dosage form, and route of administration. Therapeutic equivalence means they produce the same clinical effect and safety profile. A generic can be pharmaceutically equivalent but not therapeutically equivalent if it’s absorbed differently or acts unpredictably. Pharmacokinetic studies are designed to bridge that gap-but they don’t always succeed.
Releted Post


Andy Dargon
Hi, I'm Aiden Lockhart, a pharmaceutical expert with a passion for writing about medications and diseases. With years of experience in the pharmaceutical industry, I enjoy sharing my knowledge with others to help them make informed decisions about their health. I love researching new developments in medication and staying up-to-date with the latest advancements in disease treatment. As a writer, I strive to provide accurate, comprehensive information to my readers and contribute to raising awareness about various health conditions.
So let me get this straight-we’re trusting that a pill that can be 20% weaker OR 25% stronger than the brand is "equivalent"? That’s not science, that’s a gamble with people’s lives. And don’t even get me started on the "washout period"-what if someone’s metabolism changes during that time? Or they had a bad night’s sleep? Or ate a burrito? This whole system feels like a glorified coin flip with a PhD sticker on it.
I switched my generic antidepressant last month and honestly? I felt like a zombie for two weeks. 😔 Then I switched back to brand and boom-back to normal. Not saying generics are bad, but… my body noticed something. Maybe regulators should listen more to patients, not just lab data. 🤷♀️
Hey, I’m not a scientist, but I’ve been on generics for 15 years and never had an issue. I think most people are fine. But I get it-some drugs are tricky. Maybe we just need better testing for those few cases? No need to throw the whole system out. 😊
This is why we can’t have nice things. The FDA lets this slide because big pharma doesn’t want to spend money on real testing. And now we’re all guinea pigs. You think your blood levels are being tracked? No. You’re just a number in a spreadsheet. And your health? A cost-cutting metric.
They’re lying to you. The real reason they don’t test properly is because the Chinese factories are cutting corners-and the FDA is in bed with them. I’ve seen the documents. They’re using recycled fillers. The same stuff they put in dog food. And you’re swallowing it. Wake up. 🇺🇸🔥
Ha. So America spends $1M to test a pill? In India, we just check if it’s white and round. If yes, we sell it. Same pill. Same name. Same price. You think your blood levels matter? 😂
Generic drugs save lives. Stop complaining.
Wait-so you’re saying that for a drug like warfarin, two generics with identical blood levels can still cause different outcomes? That’s wild. And yet, we still let pharmacists switch them without telling you? That’s not just lazy-that’s dangerous. I’ve seen people get INR spikes after a switch. And no one’s tracking it. Why? Because no one’s looking. And if no one’s looking… who’s responsible?